最严格监管时代医疗器械行业该如何应对
国家食品药品监督管理总局(CFDA)在2015年最后一个长假前又发布了一份重要通知——《医疗器械生产质量管理规范现场检查指导原则》(以下简称《指导原则》),让医疗器械圈的RA人员、质量管理体系工程师及公司的老板们度过了一个纠结的假期,引发了圈内的广泛讨论。
检查结论及其判定标准:
1、污僻莆姆产品注册和生产许可“整改后复查”的企业应当在现场检查结束后的规定时限内[其中注册核查在6个月内,生产许可(含变更)现场检查在30天内]完成整观鲼视防改并向原审查部门一次性提交整改报告,审 查部门必要时可安排进行现场复查,全部项目符合要求的,建议结论为“通过检查”。对于规定时限内未能提交整改报告或复查仍存在不符合项目的,建议结论为 “未通过检查”。 在产品注册时意味着产品注册申请将不予通过。在生产许可延续申请时,将不予批准延续。企业将会丧失产品生产、销售的资格。
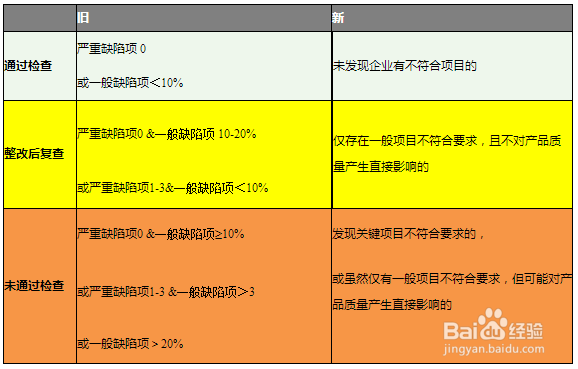
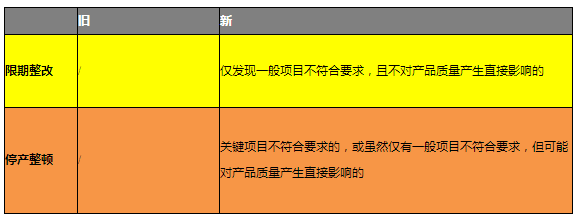
2、监娴捱颐锑督检查检查指导原则明确了限期整改和停产整顿的条件,为650号令第67条及局令第7号中相关条款提供了解释,为执法提供了依据。很多公司,在首次注册时花大力气建立质量管失窝蜜琶理体系,然而取得注册证后就没再按质量体系的要求组织产品生产,这些企业将会在日常监督检查中面临巨大的考验,应尽早改变想法,维持并完善质量管理体系。
二、检查时机与执行人员
1、产品注册 新法规要求在注册审评过程中进行体系核查,并且注册审评老师可能参与到现场审核中,核查重点处理包含注册审评老师,除了对人员、硬件、样品生产真实性进行核查外,会突出对设计开发记录与注册资料一致性的核查。
2、生产许可 在生产许可变更及延续时,由市局按“规范”进行核查。
3、监督检查 除了每年定期的日常监督检查外,企业还可能会被国家局和省局进行飞行检查。以往现场检查后通常是提出不符合项,要求企业整改,而现在企业存在不符合情况时,可能会被要求停产整顿。
三、主要关注点
1、风险管理 更加强调了全过程的风险管理。风险管理的输出与质量管理体系活动是否一致。
2、人员 企业负责人、管理者代表、检验员及人员培训的相关条款作为重点项。要求企业负责人、管理者代表应该提高法规意识,熟悉法律法规并应用到企业管理中,同时很多省份还出台了“管理者代表管理办法”,管理者代表被认为是医疗器械行业最尴尬的职位,这值得企业深思。
3、厂房、设施、设备 根据产品生产规模、生产工艺进行合理配置,注重日常管理、维护、校准等工作。
4、文件 将产品技术文件(产品主文档)的要求作为重点项,这是产品生产制造的依据。应保证批记录应与之对应。
5、设计开发 明确了“提交给注册审批部门的文件”作为“设计输出”的一部分,并且作为核查重点项。要求企业在进行产品设计开发时应考虑并实现相关法规和标准的要求,保证注册资料与设计开发原始记录的一致性。同时,明确了设计变更的控制的要求,进行风险分析,采取控制措施降低风险到可接受水平。
6、采购及生产过程控制 突出了“可追溯”的要求,这也是大部分企业在体系核查中经常被开具不符合的条款。如何定义产品批号、如何组成生产批、追溯程度和追溯范围如何定义,这些都应该进行详细策划,并满足法规要求。 医疗器械企业大多是小微企业,在供应商处的话语权也不够,但对原材料的要求又搞,所以很多企业无法与供应商签署“质量协议”,这也是企业常遇到的难题。在指导原则的要求中,一旦某个重要物料没有“质量协议”,将面临“不予注册”或“停产整顿”的风险。
7、质量控制及不合格品控制 产品应该满足“强制性标准”或“技术要求”的性能指标。已经拿到注册证的企业应该自查企业的检验设备、检验能力是否满足要求,并考虑在产品生产的适当时机完成相关测试。
8、不良事件监测、产品召回 企业应当按规定进行不良事件监测、汇报和产品召回工作,否则不仅在体系核查是会被开具不符合,在产品延续注册时也会遇到很大困难,可能导致无法延续。